Science专刊论文解读:生物多样性保护与人类健康新启示
长期以来,非人灵长类动物基因组研究帮助我们更好地了解了人类进化起源、健康和疾病。然而过去研究仅针对少数关键灵长类谱系的基因组,例如类人猿或猕猴,十分有限。同时当前气候变化、栖息地丧失及非法贸易和狩猎导致的灭绝危机,世界上60%的灵长类物种面临灭绝风险,因此也非常迫切需要全面了解野生灵长类动物的遗传多样性及其决定因素,为生物多样性保护提供指导。近日,多家国际团队联合构建了迄今为止最丰富的灵长类物种基因组数据集,并以此展开了分析该,解决了一系列相关问题,该研究是灵长类基因组计划的扩展项目成果之一,以“A global catalog of whole-genome diversity from 233 primate species”(来自233种灵长类动物的全基因组多样性全球目录)为题的论文,在著名学术期刊《科学》(Science)发表。
最丰富的灵长类物种基因组数据集
由托马斯·马奎斯-博内特(Tomàs Marquès-Bonet)教授领衔的多家国际团队联合对来自211个灵长类动物的703个个体基因组进行了高深度测序。综合先前发表的29个物种共计106个个体数据,构建了目前多样性最丰富的灵长类物种基因组数据集,涵盖了灵长类动物全部的科、86%的属和47%的物种。
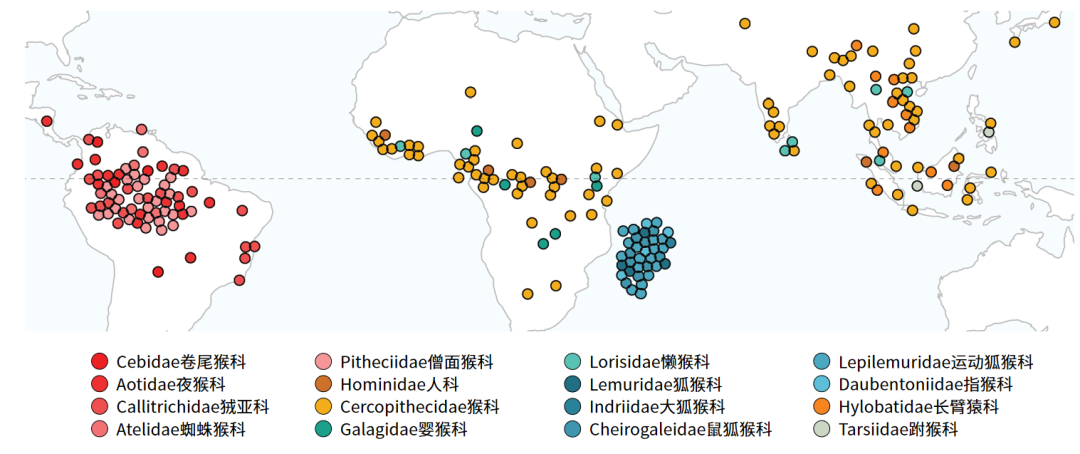
图1 项目所采集的非人灵长类物种分布点(Credit: Lukas Kuderna)
在全部样本中,超过72%的个体来自野外,58%的物种被世界自然保护联盟IUCN归类为濒临灭绝。如西黑冠长臂猿(Nomascus concolor)仅估计有1500个个体散布在一系列不连续的野外栖息地,再如北鼬狐猴(Lepilemur septentrionalis)估计大约仅有40个个体生活在野外。在空间范围上,研究样本涵盖了包括美洲、非洲大陆、马达加斯加和亚洲等目前非人灵长类动物栖息的所有主要地理区域。
重构精度更高的灵长类系统发育关系
覆盖度更大的基因组数据为重构精度更高的灵长类物种演化关系提供了基础。结合化石证据,该项目利用超保守元件(UCE)及其侧翼区域序列构建灵长类物种全基因组核系统发育关系(图2)。简鼻亚目(Haplorhini)和原猴亚目(Strepsirrhini)之间的分歧发生在63.3-58.3百万年前,因此灵长类冠群的辐射适应完全发生在古新世。眼镜猴内部最深的分歧最近出现在15.2-9.5百万年前,暗示沿着导致现存跗猴的长分支曾发生过相当大的灭绝事件。
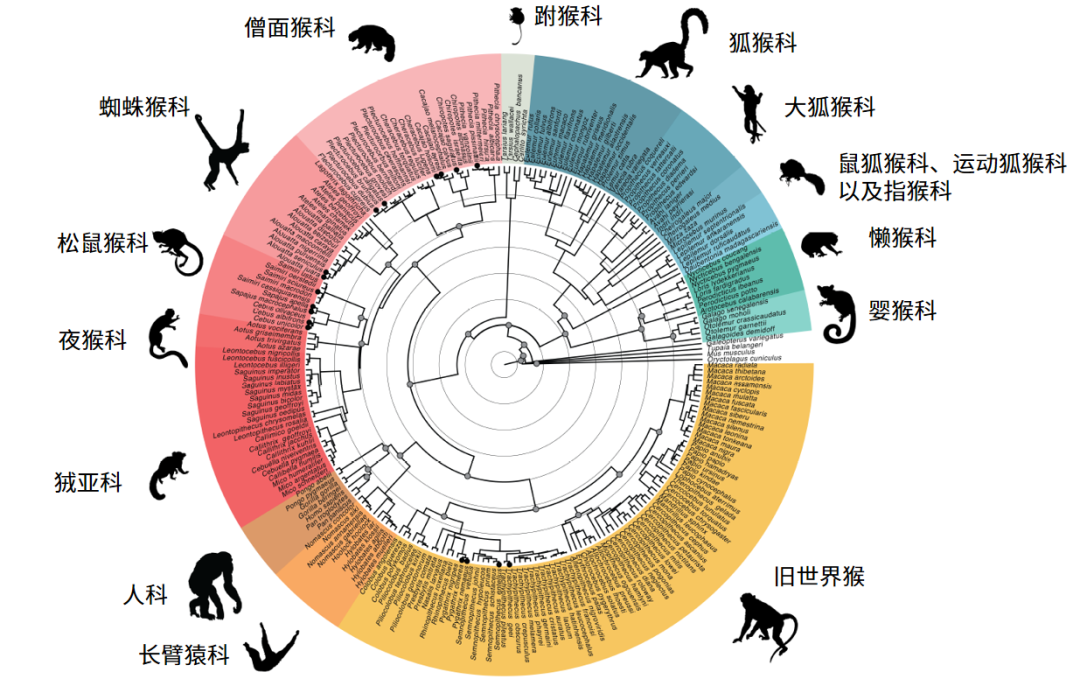
图2 化石校准的灵长类系统发育时间树。内部节点实心灰色圆圈表示化石校准点,尖端实心圆圈标记的物种表示存在并系或多系情况(Credit: Lukas Kuderna)
人类与黑猩猩之间的分化时间在900至690万年前之间,这比最近其他分析得出的时间稍早。值得一提的是,来自17个物种中的同种个体在拓扑树上并没有严格聚类在一起,即存在并系或多系分布(图2),该结果对目前建立的几个物种边界提出了质疑。这些案例可能是由于物种划界、不完全谱系分选或者杂交等。
基因组杂合性是否可以衡量物种濒危程度?
基因组杂合性可以一定程度上反映一个物种的遗传多态性。灵长类物种基因组杂合性跨度大,平均一千个碱基有0.41到7.14个杂合位点。川金丝猴(Rhinopithecus roxellana)具有最低水平多样性,大约每2400个碱基有一个杂合位点。有趣的是,只有15个物种的遗传多样性低于人类,包括几种亚洲疣猴、指猴、西白毛长臂猿和几内亚狒狒。此外,跨属、科和地理区域的遗传多样性之间也存在显著差异。
遗传多样性是否与灭绝风险相关,这是一个一直被讨论的话题,这决定了遗传多态性能否作为衡量物种灭绝风险的指标之一。这项研究表明世界自然保护联盟所划分的灭绝风险类别与灵长类基因组杂合性之间没有显著关系(图3A)。由于遗传多样性在很大程度上取决于长期的种群动态历史,因此在跨物种比较中不太可能检测到许多灵长类动物目前经历的近期种群大小的快速下降。然而比较同一科内未受威胁和受威胁物种的遗传多样性发现,受威胁的物种的多样性较低,尽管并非所有比较都达到统计显着性(图3B)。唯一的例外是懒猴科(Lorisidae),它在未受威胁和受威胁物种之间的遗传多样性上没有差异。
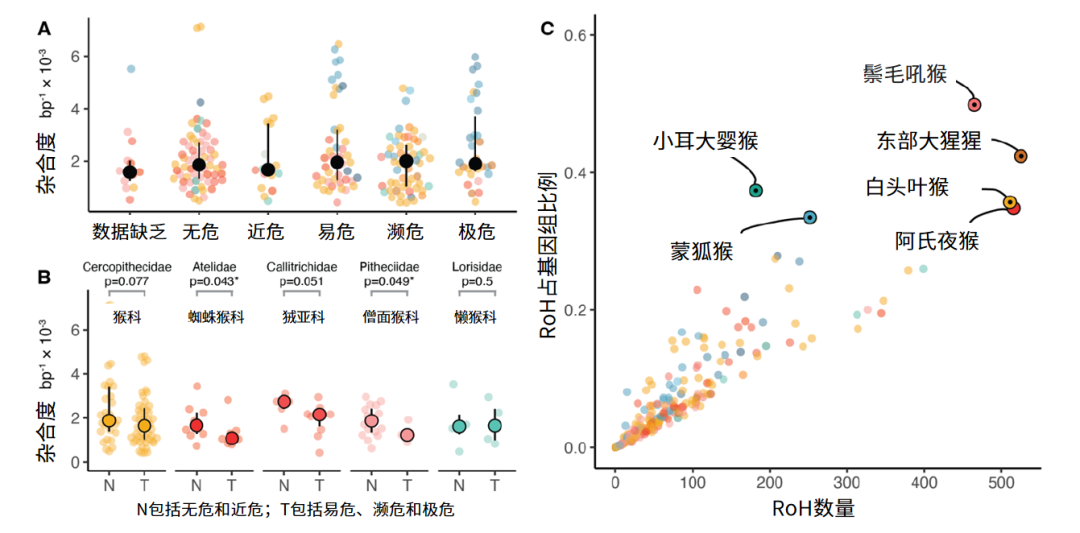
图3 灵长类基因组杂合度与物种濒危程度(Credit: Lukas Kuderna)
纯合性片段(RoH)也是衡量基因组杂合性的指标之一。该项目发现全基因组范围内RoH比例最高的物种中存在极危物种,如白头叶猴(Trachypithecus leucocephalus)、东部大猩猩(Gorilla beringei)和蒙狐猴(Eulemur mongoz)(图3C)。然而,一些目前未被归类为濒危的物种,例如阿氏夜猴(Aotus azarae)和小耳大婴猴(Otolemur garnettii),也有很大一部分基因组处于RoH状态。虽然这两个物种的整体保护状况可能并不令人担忧,但有些个体可能来自较小的当地种群,这可能会加剧近亲繁殖。重要的是,灭绝风险与基于RoH全基因组比例推断的近亲繁殖程度之间没有显著关系,这意味着RoH也不能很好地反映灵长类动物灭绝风险。许多极度濒危的物种受到非遗传因素的威胁,尽管种群数量下降速度很快,但无法在基因组水平上得到及时体现。
漂移屏障假说在灵长类中获得支持
漂移屏障假说认为代突变率(μ)会随着有效种群大小(Ne)的增加而减少,因为影响适应性的新突变主要是有害的,并且选择较低突变率的能力随着种群规模的增加而增加。
本研究估计灵长类动物的世代突变率在0.25×10-8和1.62×10-8之间变化,这比之前报道的范围大得多,其中狐猴科具有最低的突变率,人科具有最高的突变率。此外还发现,代突变率与世代时间之间存在显著正相关关系(图4A),这部分抵消了世代时间对年突变率的影响。因此,后者年突变率在世代时间较短的物种中更大(图4B)。
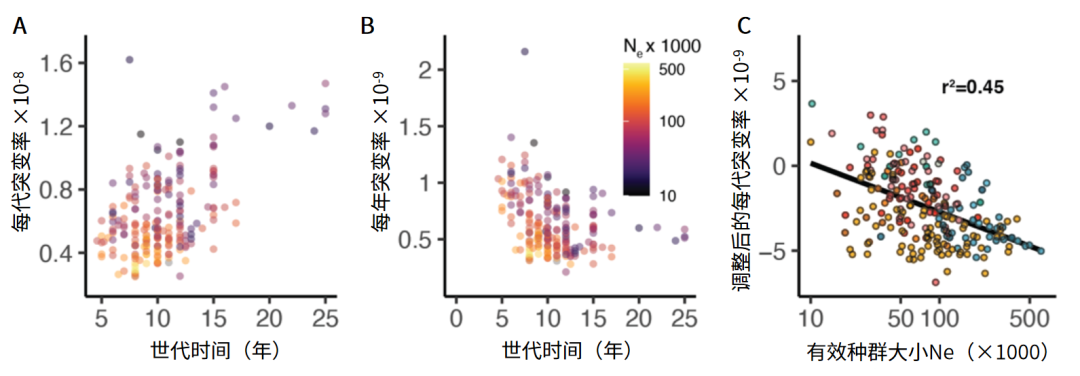
图4 灵长类物种基因组突变率与有效种群大小(Credit: Lukas Kuderna)
研究发现长尾猴属和美狐猴属的多个成员表现出高Ne值,这可能是由种间杂交导致的。类人猿、懒猴和阔鼻类动物的有效种群大小相对较低。该项目还发现有效种群大小高的物种的突变率较小(图4C),这支持了漂移屏障假说。
影响灵长类遗传多样性的新因素
为了进一步弄清楚哪些因素可能会影响遗传多样性和突变率,项目基于系统发育广义最小二乘法(PGLS)模型,使用遗传多样性或突变率作为响应变量,并将个体性状作为预测因子,分析了32种物种特征与多样性或突变率之间的关系。
研究发现,交配系统、活动预算(简单点来说是指,动物在进食、休息、睡眠和移动等各种活动中花费的时间)、气候生态位、分布模式等特征是多样性的重要预测因子。比如,单一雄性一夫多妻制交配系统中的物种表现更低的多样性,这与雄性等位基因多样性贡献减少的预期一致。在气候生态位内,多样性梯度从南到北下降,这是由南半球高度多样化的狐猴物种驱动的。此外还发现多样性与平均温度和降水量显着相关,但体重或寿命等生活史特征和灵长类动物的遗传多样性之间没有显著关系。
灵长类基因组变异图谱揭示人类性状与疾病基础
该研究更新了之前发布的647种人类特异性高频错义突变数据集,即在人类谱系中特异性出现并迅速上升到高频或固定的氨基酸改变变异。虽然这些变异不足以完全解释人类的独特性,但这样的变异集应该包含其某些分子基础。最新研究发现63%的人类高频特异性错义突变发生在至少一种其他灵长类动物中,55%发生在超过两种灵长类动物中,这表明同一个突变的复发可能在灵长类动物中普遍存在。
研究进一步检测哪些基因在其他灵长类物种中没有表现出频繁的等位基因复发,最后发现了影响了80个基因的89个错义突变。发现了人类和尼安德特人之间已证明功能差异的两种氨基酸差异:NOVA1(神经肿瘤腹侧抗原1)中的祖先等位基因导致皮质类器官发育缓慢并改变突触蛋白相互作用,以及腺苷酸琥珀酸裂解酶基因(ADSL)的人源等位基因导致大脑中嘌呤的从头合成减少。此外,先前报道的有丝分裂纺锤体相关基因(SPAG5、KIF18A)的变异是人类特有的,这可能影响了发育过程中的神经发生。有趣的是,基因TMPRSS2中发现了一种人类特有的变异,这是对新冠病毒感染做出反应的主要因素。
该研究还构建了一个包含近百万个位点的类人猿谱系特有突变。这些突变包含影响了2970个基因的3792个错义突变,这些基因显著富集在多个与纤毛相关的功能类别,暗示了类人猿特异性纤毛进化对于塑造人类谱系很重要。正常功能纤毛的破坏会导致人类出现一系列异质性病理,统称纤毛病。在与纤毛病相关的187个基因中,有30%受到类人猿特异性错义突变的影响。这表明在某种程度上引起猿类特异性表型的突变最终也可能会影响人类表型。








