Science专刊论文解读:用灵长类动物基因数据训练AI识别致病性基因突变
基因突变是许多疾病产生的重要原因,但我们对这些基因突变致病性的理解仍然非常有限。随着个体基因组测序的普及,已经揭示了个体之间存在数百万种基因差异,但我们对它们的临床意义的理解仍然非常有限。Illumina人工智能实验室的首席研究员Kyle Kai-How Farh领导的联合研究团队通过灵长类动物基因数据集训练了一个名为PrimateAI-3D的人工智能神经网络,为解释人类基因变异的致病性找到了新方法,对精准医学具有重大意义。作为灵长类基因组计划的国际扩展项目,近日,该项研究相关成果已经以题为“The landscape of tolerated genetic variation in humans and primates”(人类和灵长类群体中可容忍的遗传变异景观)的论文发表在了著名的学术期刊《科学》(Science)上。
精准医学需要识别致病性基因变异
精准医学是一种根据患者内在的生物学信息和临床症状等,为患者提供个性化的预防、诊断和治疗方案的医学模式。随着人类基因组计划(HGP)的完成以及测序技术的发展,生物信息学数据量急剧增长,我们已经揭示了个体之间大量的基因变异。准确识别人类基因变异的致病性能够促进精准医学的发展。通过对人类基因变异的识别和功能的阐明,研究人员能够找出许多特定基因突变与疾病的关系。这样当我们获得一个人体内的所有基因变异,就可以评估这些变异导致的疾病风险,从而采取相应的预防、治疗措施。
然而,在人类基因组中,可能导致蛋白质改变的基因变异数量超过了7000万种,其中仅约0.1%的变异在临床变异数据库(如ClinVar)中被注释,其余变异的临床意义并不明确。尽管科学界合力研究,但大多数人类基因变异的罕见性使得破译个人基因组变异的意义进展缓慢。如何识别这些变异的致病性仍然是一个艰巨的挑战。
也正是因为如此,对患者进行的临床测序测试结果经常会缺乏明确的诊断指导意义,甚至可能给出错误的诊断指导建议,这会让患者和临床医生都感到沮丧。甚至在某些情况下,当假定为致病突变的基因型后来会被发现是以前未被研究的人群中的常见变异,结果往往还需要重新联系病人并推翻之前的诊断——因为常见变异一般不会导致严重的遗传病,自然选择会把导致严重疾病的基因压制在很低的频率上(当然也有一些特殊情况,比如创始效应(founder effects)和平衡选择会导致一些罕见的例外情况)。
为了实现精准遗传诊断,我们必须能够准确地从所有人类基因组中存在的广泛基因变异中,将真正的致病或与疾病相关的基因变异鉴定出来,因此迫切需要一种可扩展的方法来解释人类基因变异的影响及其对疾病风险的贡献。
为什么研究灵长类动物基因组
灵长类动物是与人类亲缘关系最近的动物类群。如黑猩猩和人类的蛋白质序列相似度高达99.4%,这表明在黑猩猩中发现的蛋白质变异很可能会在人类中具有同样或接近的效果。我们可以通过对与人类密切相关的灵长类动物进行群体测序研究,来系统地分析常见变异——这些常见的变异在灵长类中很可能是无害或者低害的。而与本研究同时发表的另一篇研究成果显示,同一个突变的复发可能在灵长类动物中普遍存在。这就意味着在其他灵长类上发生的基因突变可能在人类中同样存在类似的情况,而在其在灵长类中正常个体所表现出的低致病性可以用来排除它们在人体内的致病性。这就像通过测序更多多样化的人类群体来推进临床对变异的解释一样。
然而,在早期的研究中,可用的灵长类动物群体测序数据非常有限,这限制了发现的常见变异的数量,也限制了机器学习分类器的规模。因此,如果我们能够构建更大规模的灵长类动物基因数据集,并进一步提高机器学习算法的准确度和规模,那么这种方法有望成为解决问题的可行方案,为精准医学的发展提供更好的支持。
构建包含430万个灵长类动物良性错义变体的数据库
为了扩展可用的灵长类动物基因数据集,研究人员对211种灵长类动物的703个个体进行了测序,并结合之前的研究数据,总共包括了233种物种的809个个体。这些物种覆盖了地球现存的521种灵长类动物的一半,涵盖了所有主要的灵长类动物科,包括旧世界猴 (Old World monkeys)、新世界猴 (New World monkeys)、狐猴 (lemurs) 和眼镜猴 (tarsiers) 等。研究人员选择了每种物种的少数个体(平均3.5个)进行测序,以过滤掉罕见的变异。研究发现,这些灵长类动物中存在着430万个独特的错义(改变氨基酸)变异和670万个独特的同义(无法改变氨基酸)变异。
研究人员将灵长类动物变异数据与人类基因组聚合数据库(gnomAD)进行比较后发现,有30%的可能的同义变异重合,而可能的错义变异只有6%重合。这些检测到的常见的错义变异在灵长类动物种群中以高频率出现,它们在经历了自然选择的筛选后仍然能生存下来,表明不太可能会致病。
另外,进一步研究表明,在灵长类动物中发现的错义变异与ClinVar临床变异数据库中的已知良性突变具有很高的一致性。在审查致病级别较高(两星及以上,表明多个提交者达成共识)的ClinVar变异中,有99%的错义变异被认为是良性或可能良性的。本研究公开的灵长类群体常见错义变体数据库包含了超过430万个可能是良性的错义变体,可供基因组学研究参考。对于注释的错义变体数量而言,该数据库规模比ClinVar数据库大50倍,且几乎完全由先前未知意义的变体组成。比较这些灵长类动物变体与人类变体,有助于揭示自然选择作用下的基因突变模式,为临床诊断提供了有价值的参考。
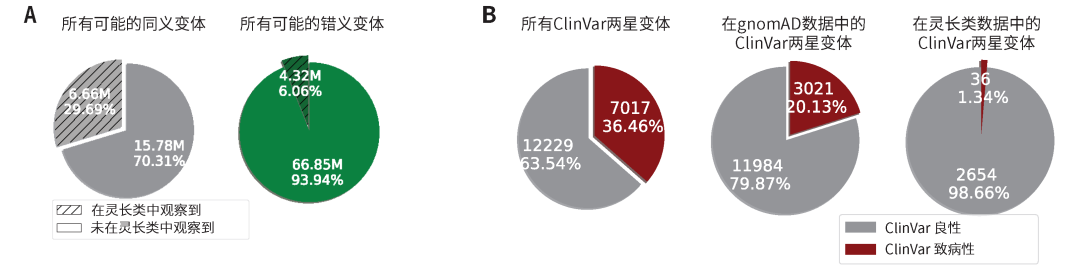
灵长类动物常见变体与已知人类数据库的比较。A, 在灵长类动物中观察到的所有可能的人类同义变体(灰色)和错义变体(绿色)的部分;B, 在整个ClinVar数据库中具有双星评审状态或以上的良性(灰色)和致病性(红色)错义变体的统计(左),在gnomAD中观察到的ClinVar变体(中),在灵长类动物中观察到ClinVar变体(右)(Kyle Farh (Illumina))
人类与灵长类动物的基因水平选择约束
选择约束(selective constraint)是指在纯化选择(purifying selection)的作用下,相比于中性演化序列,功能序列的演化分歧较少的现象。研究人员通过比较人类基因与灵长类动物整体的选择约束,发现单个基因的错义突变和同义突变的比率在人类和灵长类之间具有很好的相关性(Spearman r = 0.637),这表明在人类中因有害错义突变而缺失的基因在整个灵长类动物谱系中也普遍缺失。此外,研究人员还发现,人类和灵长类动物的基因错义突变和同义突变的比例与基因失活不耐受概率(probability of genes being loss of function intolerant, pLI)的相关性也相似(Spearman 相关系数分别为 -0.534和 -0.489)。
为了衡量每个基因的选择约束力,研究人员使用三核苷酸突变率来模拟每个变异体的期望观察概率,并计算每个基因的实际观察值与期望值之间的关系。他们发现,在人类和灵长类动物中,同义突变的实际观察值和期望值高度相关,但是错义突变的实际观察值和期望值存在明显差异。这可能是因为高度约束的基因(如高pLI基因)中致命的错义变异会被自然选择清除掉。在灵长类动物中,高度约束的基因中的错义变异几乎完全被清除了,而在gnomAD中,由于其大样本量,罕见的错义变异几乎没被清除。
接着,研究人员使用了两种方法(群体遗传建模和泊松广义线性混合模型)来确定选择约束在人类和灵长类动物中的差异,并取两种方法的交集。结果发现,有39个基因在人类和灵长类动物中的选择性约束存在显著差异(Benjamini-Hochberg FDR < 0.05)。其中,相对于灵长类动物,人类选择性约束降低最多的三个基因是CFTR、GJB2和CD36,它们是囊性纤维化(cystic fibrosis)、遗传性耳聋(hereditary deafness)和血小板糖蛋白缺乏症(platelet glycoprotein deficiency)的常染色体隐性遗传基因。这些基因中的致病突变在特定地理人群中异常普遍,可能是由杂合子优势导致的,这种杂合子优势可以保护人体免受特定环境病原体的伤害。
另外,研究人员还发现人类中TERT基因的选择约束显著增加,这个基因在维持端粒长度方面起着重要作用,可能与人类比其他灵长类物种更长寿有关。需要指出的是,由于灵长类动物的样本数较少,目前研究人员还无法确定这种选择约束的增加是否只在人类中出现,亦或是在大猩猩等其他灵长类动物演化历程中就已开始。通过增加灵长类动物的样本数量,将可以更好地检测物种特异性和谱系特异性的演化适应,并揭示导致现代人类状态的演化路径。
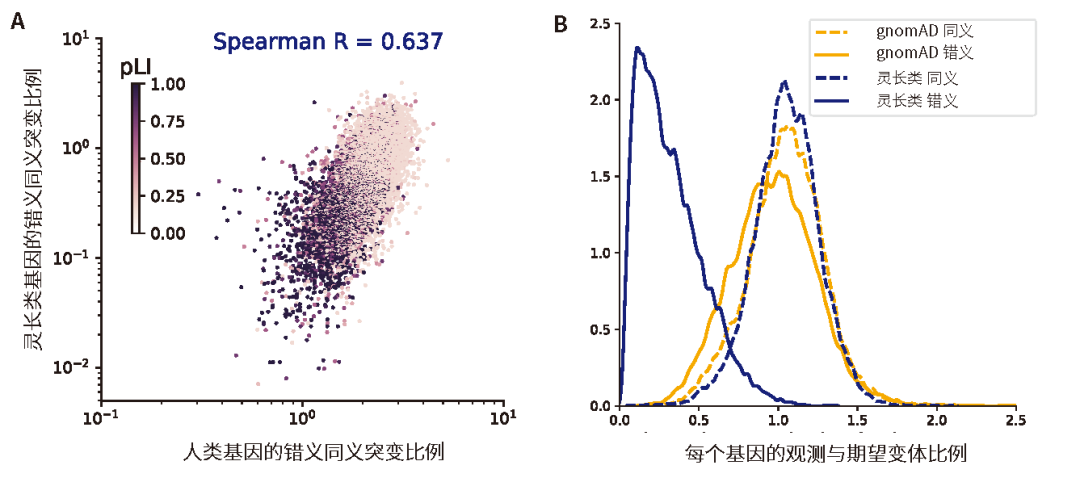
人类与灵长类基因的选择性约束比较。A, 灵长类动物和人类基因之间的错义同义突变比率的散点图。每个基因都由其 pLI 分数着色,颜色较深的点显示单倍计量不足基因。;B, 所有基因的同义(虚线)和错义(实线)变体的观察/预期比率的分布。显示了灵长类动物基因(橙色)和gnomAD基因(蓝色)的结果。(Kyle Farh (Illumina))
PrimateAI-3D,一个用于预测错义变异致病性的深度学习网络
研究人员构建了用于变异致病性预测的半监督的三维卷积神经网络PrimateAI-3D。与以往在线性序列上操作的深度学习架构不同,PrimateAI-3D将蛋白质的三维结构在2埃分辨率下进行体素化,并使用三维卷积来使得网络能够准确地识别仅从序列上看不出差异的关键结构区域,从而更准确地预测变异的致病性。例如,STK11的PrimateAI-3D预测结果展示,每个氨基酸位置由该位置的平均PrimateAI-3D评分着色,该基因与Peutz-Jeghers遗传性息肉病综合征相关。在训练中,灵长类动物常见变异和注释的ClinVar致病性变异来自线性序列的不同部分,在三维空间中形成不同的聚类。值得注意的是,这个网络没有使用人工设计的特征或在临床变异数据库中注释的变异进行训练,从而避免了变异注释中的人为偏见。相反,它基于灵长类动物常见变体的局部富集或减少来推断致病性,仅采用蛋白质的多序列比对和三维结构作为输入。
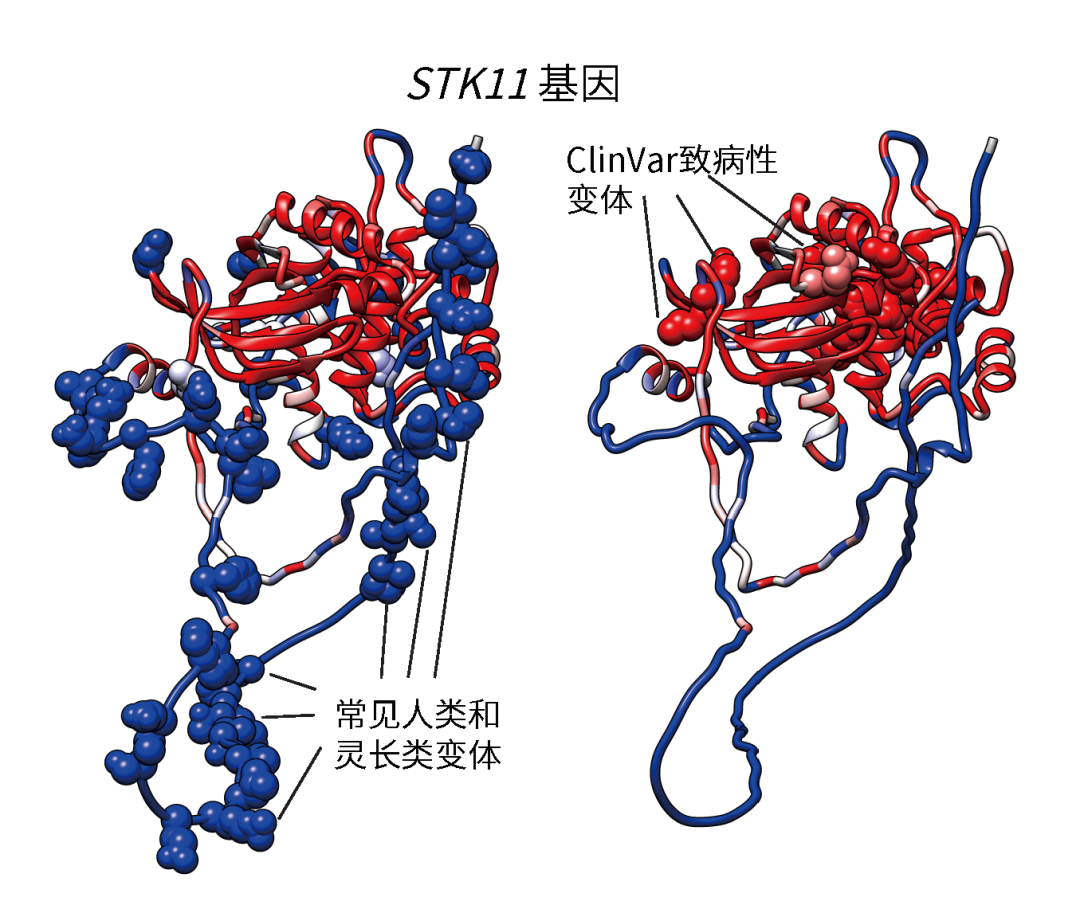
STK11基因的蛋白质结构,由PrimateAI-3D致病性预测评分着色(蓝色:良性;红色:致病性)。球体表示具有常见人类和灵长类变体的残基(左)或具有ClinVar致病突变的残基(右)。对于球体,颜色仅对应于变异的致病性评分。对于其他残基,致病性得分是该位点所有变异的平均值(Kyle Farh (Illumina))
PrimateAI-3D可利用实验或计算预测得到的蛋白质结构,研究人员使用AlphaFold DB和HHpred预测的结构以覆盖尽可能广泛的人类基因。对于训练数据,研究人员整合了来自233种灵长类动物的所有常见错义变体和gnomAD、TOPMed和UK Biobank中的人类常见错义变体(等位基因频率>0.1%的人群),共计450万个可能是良性结果的错义变异。该数据集涵盖了所有可能的人类错义变异的6.34%,是当前ClinVar数据库(在排除不确定意义的变异和具有冲突注释的变异后,有79,381个错义变异)的50倍以上,极度扩大了机器学习可用的训练数据集。由于训练数据集仅包含标记为良性的变异,研究人员创建了一组随机选择的变异对照,这些变异通过三核苷酸突变率与常见变异相匹配,并训练PrimateAI-3D将常见变异从匹配的对照组中分离出来,作为一项半监督学习任务。
在进行变异分类任务的同时,研究人员借鉴了为了”预测句子中缺失单词“的语言模型架构,为蛋白质中的每个位置生成了氨基酸替换概率,方法是遮盖并使用序列上下文来预测缺失的氨基酸。研究人员为此训练了两个模型:
1)一个三维卷积“填空”模型,它的任务是预测体素化的三维蛋白质结构中缺失的氨基酸;
2)一个使用transformer架构来利用周围多序列比对作为上下文预测缺失氨基酸的语言模型。
作者将这些模型作为额外的损失函数,以进一步优化PrimateAI-3D的预测结果。研究人员还训练了一个多序列比对的变分自编码器(variational autoencoder, VAE),并发现它的性能与他们的transformer架构相当。因此,研究人员将它们的预测结果平均值纳入损失函数中,这比单独使用任何一个模型都要好。
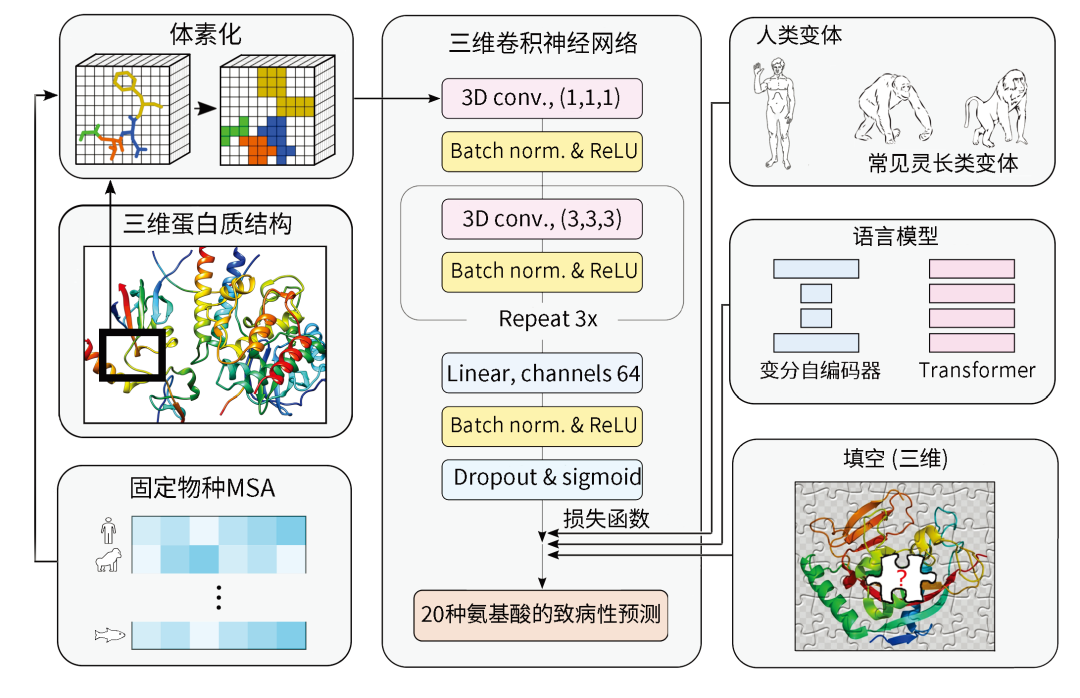
PrimateAI-3D工作流程。将人类蛋白质结构和多序列比对体素化(左)作为3D卷积神经网络的输入,该网络预测目标残基的所有可能的点突变的致病性(中)。该网络是使用具有三个组成部分的损失函数进行训练的(右):常见的人类和灵长类变体;填充蛋白质结构的空白;基于语言模型的分数排名(Kyle Farh (Illumina))
研究人员评估了PrimateAI-3D和其他15种已发布的机器学习方法,展示了它们在六个不同数据集上区分良性和致病性变异的能力。在测试的四个患者队列 (UKBB, DDD, ASD, CHD) 中,PrimateAI-3D在区分致病性和良性变异方面优于所有其他分类器;在ClinVar注释数据库中,它在分离致病性和良性变异方面表现也是最好的,并且与体外深度突变扫描分析 (DMS assays) 具有最高的平均相关性。在PrimateAI-3D之后,没有明显的亚军,在六个不同的基准中,六个不同分类器占据了第二名。值得注意的是,PrimateAI-3D在临床变异基准上的表现与训练数据集的大小直接相关。因此,额外的灵长类测序数据将是解锁更多收益的关键。
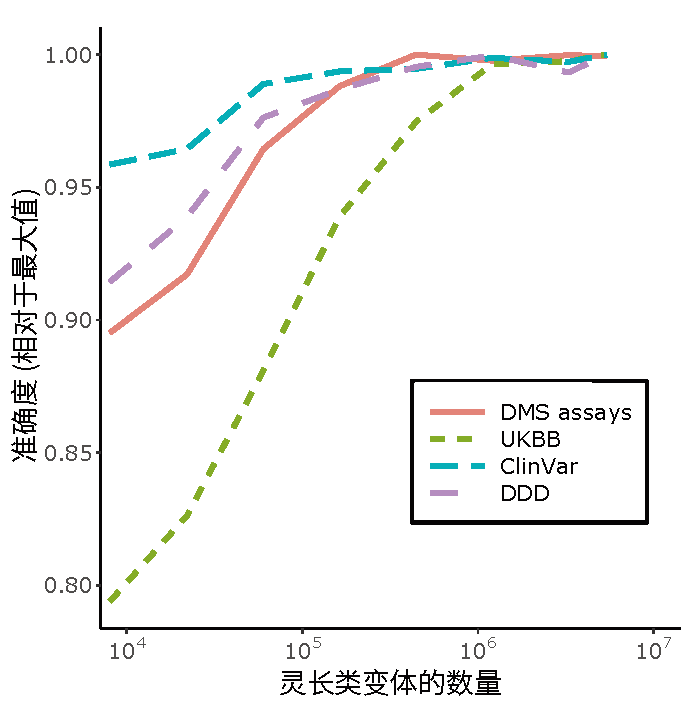
训练数据集大小对分类精度的影响。随着训练数据集中常见灵长类变异数量(x轴)的增加,PrimateAI-3D 的性能得到改善。每个数据集的性能为所有训练数据集大小中观察到的最大性能的相对值(y 轴)。(Kyle Farh (Illumina))
发现神经发育障碍的候选疾病基因
新生突变(de novo mutation)是指在精卵细胞结合或受精卵发育过程中自发产生的一种突变,其在父母的基因组中并不存在。虽然每个人的基因组中都有新生突变的存在,但是致病性新生突变在人群中的发生率很低。而由于致病性新生突变频率低,演化选择的压力不太严格,因此更容易产生致病性较强的突变,这使得新生突变成为散发性、复杂性疾病的主要原因之一。研究表明,新生突变在神经发育疾病中起着重要作用,例如智力障碍(ID)、自闭症谱系障碍(ASDs)。由于新生突变的稀有和复杂性,准确识别新生突变的潜在致病性可以帮助确定疾病的遗传风险和遗传模式,提供早期诊断和治疗。
研究人员使用PrimateAI-3D提高了寻找致病性新生突变富集的候选疾病基因的能力。基于三核苷酸上下文的背景突变率估计,在神经发育障碍队列DDD中受影响个体的新生错义突变比预期高了1.36倍。研究人员将PrimateAI-3D的分类阈值设为0.821,可找出与该队列中新生错义突变数量相同的致病性错义突变(n=7,238;图5A)。通过这个阈值对错义突变进行分层,将致病性新生错义突变的富集率提高到2倍(图5B),从而大大提高了发现疾病基因的能力。研究人员还应用PrimateAI-3D对致病性错义变体进行排序,发现了290个与智力障碍相关的基因,其中272个是之前已经发现的基因。
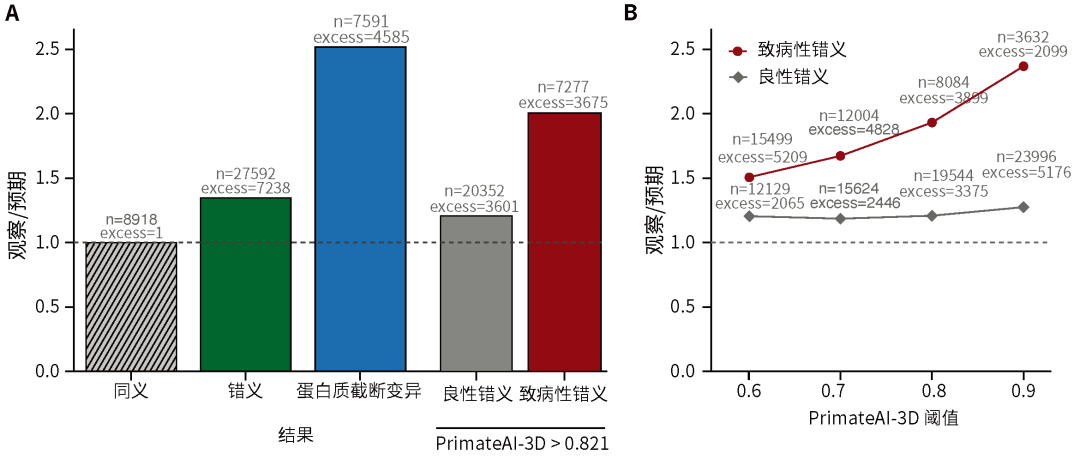
神经发育障碍队列中新生突变的富集程度超过预期。A, 来自Kaplanis等人的在所有基因中的新生错义突变的富集。给出了同义、全错义和蛋白质截断变体(PTV)的富集率,以及根据PrimateAI-3D评分将错义分为良性(<0.821)和致病性(>0.821)的结果;B, 在不同的PrimateAI-3D阈值下,良性和致病性错义的富集高于预期。(Kyle Farh (Illumina))
综上所述,这项研究成功展示了将灵长类群体测序数据和深度学习模型相结合的应用,有助于我们了解人类基因变异的致病性,帮助个性化基因组医学在临床上提供更好的诊断指导。对于推进人类遗传变异的临床理解而言,我们不能仅依赖于人类的基因变异数据,也需要探索灵长类动物的基因变异数据,这两者是互补的。利用灵长类动物的常见变体进行训练的分类器,可以通过帮助区分良性和致病的罕见变异来加速这些目标发现工作。然而,我们需要认识到,由于人类活动的影响,地球上60%以上的灵长类动物将面临灭绝的威胁。因此,采取行动来保护这些珍贵物种非常必要,这样我们可以继续从它们的基因组中汲取知识,更好地理解人类自己。








