Science专刊论文解读:人工智能,通过罕见的突变找到到容易患病的人
我们平均每个人都携带了几十个潜在有害罕见变异1,当然,还有更多的影响一般的常见遗传变异。那么,对一个人的如糖尿病和心血管疾病等疾病遗传易感性,是用数千种影响一般的常见遗传变异的总和,还是少数影响严重的罕见变异的总和来解释更好呢?Illumina人工智能实验室Kyle Kai-How Farh团队联合多家单位,使用了新近开发的PrimateAI-3D人工智能神经网络2,展示了如何将这些基因中的罕见变异组合成统一的遗传风险评分,用于预测患病的风险,初步窥见了普通人群进行个人基因组测序的潜在实用性,有助于加深我们对遗传疾病的认识和预防治疗策略的制定。作为灵长类基因组计划的扩展项目,近日,该研究的成果以“Rare penetrant mutations confer severe risk of common diseases”(罕见渗透性突变给常见疾病带来严重的风险)为标题发表在了著名学术期刊《科学》(Science)上。
罕见突变罕见到可以被忽略吗?
罕见突变是指在人群中非常罕见(等位基因频率AF<0.1%)的突变。罕见突变虽然在人群中十分罕见,但其影响绝不可忽视。在英国生物库中,研究人员观察到每个人平均携带2.96个罕见有害错义变异和0.97个罕见的功能缺失(LoF)变异。研究发现每个等位基因上这些罕见变异对人类表型的影响远远超过了常见变异,这些变异涉及到共893个基因,这些基因序列中既包含罕见变异研究,也包含常见变异研究,对比发现:罕见有害变异在相同位点上的影响比GWAS鉴定的常见变异平均大11.2倍。相比之下,常见突变通常在人群中的频率比较高,但对表型或疾病的发病贡献相对较低。
事实上,如果一个罕见突变对表型或某个疾病的发病有强烈的影响,即具有这个基因突变的个体大多数情况下都会表现出该表型或疾病,那么就可以称之为罕见的渗透性突变(Rare penetrant mutations),即意味着高的致病性,因此狭义上也可以称作罕见的致病性突变。
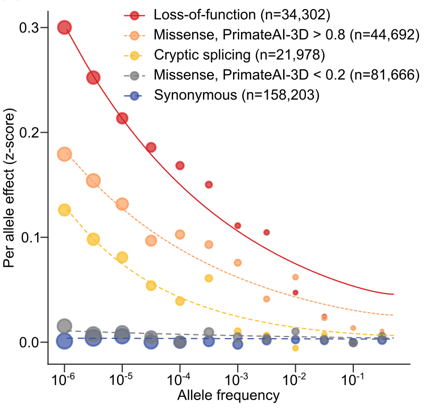
不同类别致病性突变的致病效应大小与等位基因频率之间的关系
用最罕见的突变找到最容易患病的个体
显然常见突变与表型间的关联程度往往优于罕见突变,但是在识别极端的高风险个体上也是这样吗?答案是否定的,常见变异PRS模型区分能力较弱,不能有效地识别高风险患病个体,而这些极端个体才是大部分疾病最终的病人群体。这是其在精准医学的临床应用中面临的两个最大障碍之一3。
相比之下,罕见变异会更具有优势。但是如何来识别这些与表型相关的罕见突变呢?基于灵长类基因组和人类数据共同训练出的人工智能神经网络PrimateAI-3D能够实现这个功能,它可以更好地预测人类中突变的致病性2,使得广泛理解罕见突变在人类疾病发生中扮演的角色成为可能。以PrimateAI-3D对突变的致病性的准确评估为前提,研究人员提出了一种罕见变异的多基因风险评分(polygenic risk score,PRS)系统,并使用UK Biobank的数据,构建了罕见变异PRS模型。详细地来说,首先根据关联分析确定与特定疾病相关的代谢途径、细胞类型和通路等构成的复杂网络,并找到相关的基因,再基于PrimateAI-3D对这些基因中罕见变异的致病性评分进行加权以构建计算模型,最后对于未知表型的个体,使用该模型计算其分数即可推测其患病风险。同时也构建了基于常见变异来计算得分的常见PRS模型和同时考虑两类变异的统一PRS模型作为对比。
横向来看,研究人员比较了基于其他15种方法对罕见变异进行致病性评估而构建的罕见变异PRS模型,发现基于PrimateAI-3D的PRS优于所有其他方法,表明对突变进行准确的致病性预测是罕见变异PRS高性能的前提。
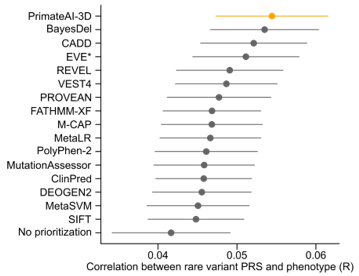
使用不同致病性分类方法构建的罕见变异PRS性能的比较
进一步地,对构建的罕见变异PRS模型、常见变异PRS模型以及两种模型的组合进行了性能测试,结果显示在78个定量表型中,统一PRS的平均表现最佳,常见变异PRS其次,均好于罕见变异PRS。这些观察结果与先前的研究结果一致,即总体而言,罕见变异解释表型的可遗传性比常见变异更少4。
尽管罕见变异PRS在平均表型预测方面较弱,但它们比常见变异PRS在识别表型极端的个体(即高患病风险个体)方面表现更好,这对于临床筛查和风险管理更为重要。表型异常值(z-score ≥3)个体与总体相比,罕见变异PRS得分位于第0.1或99.9百分位的可能性增加了10倍,是常见变异PRS的3倍,这意味着罕见变异更有可能识别出表型极端的个体。从实际上来看,例如帕金森病这一常见的神经系统变性疾病,会导致静止性震颤、运动迟缓、肌强直和姿势步态障碍,同时可伴有抑郁、便秘和睡眠障碍等非运动症状,但其确切病因仍不清楚,同时它在人群中的发病率只有0.3%5,这意味着使用罕见变异对其进行准确的预测十分重要,可以提前制定预防治疗策略,减轻疾病的影响。事实上,许多复杂的人类疾病的患病率低于1%,包括、多发性硬化症(0.3%)6、40岁前心肌梗死(0.6%)7和1型糖尿病(0.2%)8。
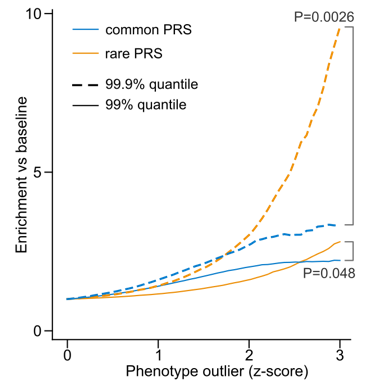
在表型异常值个体中离群值PRS分数的富集度
进一步的,研究还评估了常见和罕见PRS模型对2型糖尿病和血脂异常个体的识别能力。在疾病风险高于普通人大约4倍之前,常见变异PRS模型可以识别更多的高风险个体,而在此阈值之后,罕见变异PRS的表现更加突出。
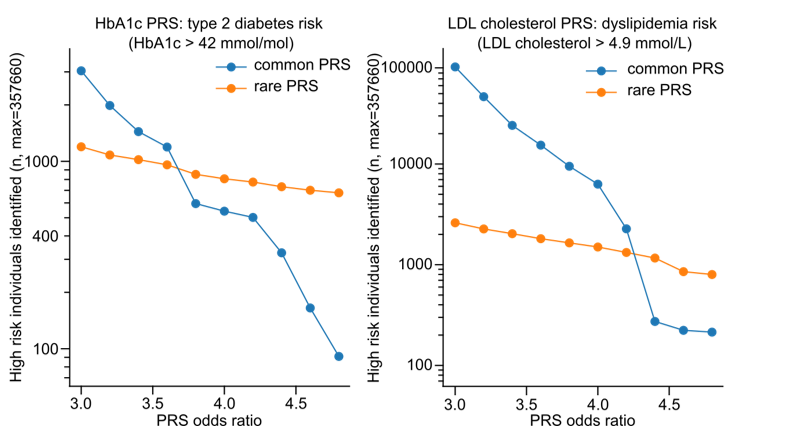
使用不同风险阈值在罕见和常见变异PRS中识别高临床风险个体的数量
综合这些发现,我们可以知道常见变异和罕见变异在预测人类疾病风险方面具有互补的效用:常见变异可以平均识别出更多可能患病的个体,而罕见变异更容易识别出最高风险的异常个体。因此将以往被忽略的罕见变异纳入PRS可能比仅使用常见变异的PRS更能识别出极端个体9,10,有助于预防性筛查。同时,这些极端个体才是大部分疾病最终的病人群体,也是最需要治疗或遭受严重的早期病变表现的群体,所以将其纳入考虑也具有更重要的临床意义11,12。
跨越种族差异:强大的可迁移性
常见变异PRS模型在不同族群之间的可迁移性有限,是其在精准医学的临床应用中面临的两个最大障碍之二3。相比之下,本研究用来构建罕见变异PRS模型的PrimateAI-3D的数据,源自233种非人类灵长类动物,这使得罕见变异PRS模型比常见变异PRS模型在迁移到其他人群方面具有优势,可以提供比目前以欧洲人为主的遗传研究更普适的结果。对此,研究人员分别评估了该模型迁移到不同数据集和不同祖先的人群中的表现。
首先将在英国生物库的欧洲个体上训练的16个罕见变异PRS模型应用于马萨诸塞州普通病人生物库(MGB)的20,708个欧洲个体,在这16个模型对应的表型中,罕见变异PRS模型的平均预测性能在两类数据中相似。值得注意的是,即使MGB数据集中有43%的罕见有害变异从未出现在用于模型训练的数据集中,罕见变异PRS模型在两个队列中却惊人地具有几乎相同的表现。因此,与常见变异PRS不同,罕见变异PRS在具有相似祖先的不同群体中具有较强的可迁移性。
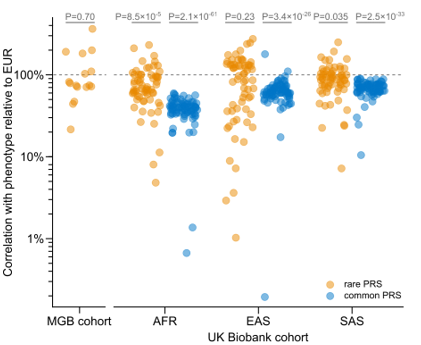
源自UK Biobank欧洲人(EUR)的罕见和常见变异PRS的表现
接下来,研究评估了罕见变异和常见变异PRS模型在仅使用欧洲人群的数据进行训练的情况下,在非欧洲人群中的表现。与以往的研究结果一致,相对于欧洲人群,在非欧洲人群中,常见和罕见变异PRS的性能的中位数均发生了下降,但罕见变异PRS表现出了更好的可迁移性。进一步的,为了评估罕见变异PRS在具有临床意义任务上的可迁移性,研究人员选择了表型分布的上下0.5%处具有PRS得分的个体,观察到欧洲人和非欧洲人在UK Biobank的测试集和MGB数据集中的两个组之间的平均表型差异相似。总体而言,使用欧洲人群数据训练的罕见变异PRS模型在测试非欧洲人群时,其在52个表型的14个表型中展现的性能优于在欧洲人群中,而常见变异PRS模型在测试非欧洲人群时表现不佳,所有52个表型的表现均低于在欧洲人群中,说明罕见变异PRS模型相较于常见变异PRS模型在具有不同祖先的群体中具有更高的可迁移性。
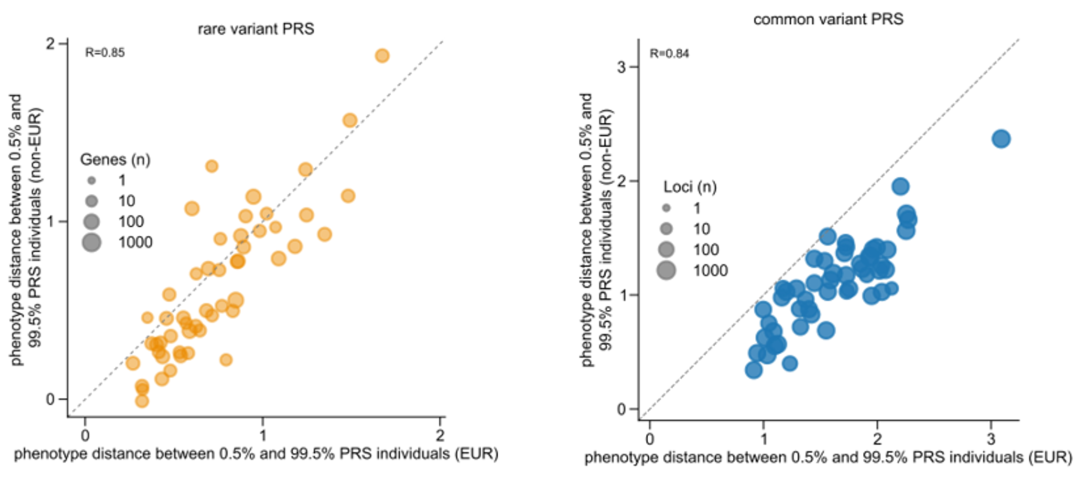
对于52种匹配的特征,显示UK Biobank EUR(x轴)和UK Biobank non-EUR(y轴)个体之间的平均表型距离。每个点代表一种表型,点的大小表示该表型相关的基因数,点落在虚线上则表示该表型对应的PRS模型在两类数据中的表现相近,在虚线上方表示在非欧洲人中表现更好,反之在欧洲人中表现更好。其中表型距离通过比较具有低(<0.5%)和高(>99.5%)罕见变异PRS百分位数的个体来计算
此外,虽然罕见突变的PRS在不同族群之间的迁移能力比常见变异的PRS更好,但它们在非欧洲人群中的平均表现仍会降低。研究表明,这可能和欧洲群体具有更准确的等位基因频率估计,以及目前人口数据库中的欧洲个体数量较多有关,导致使用欧洲人群构建的罕见变异PRS意外地包含了非欧洲人群的常见变异,从而导致其迁移后的表现下降。随着不同人群等位基因频率的估计变得更加准确和全球均一化,可以预见罕见变异PRS的可迁移性将继续改善。
更多的样本,更好的性能
研究还探讨了随着样本量的增加,罕见变异PRS性能可以提高多少。首先,关注于在识别额外显著关联基因方面的能力,研究人员在UK Biobank中进行了关联测试,观察到每个表型显著关联的基因数量随样本大小线性增加。
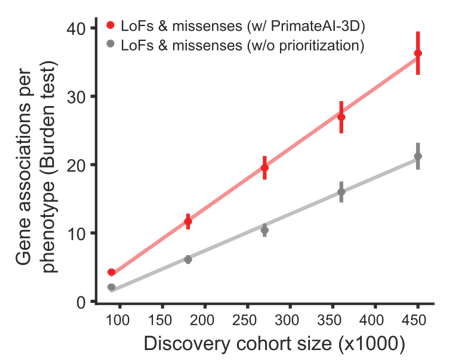
通过罕见变异负荷检测每个表型显著相关的基因数
此外,与表型关联基因的检测结果一致,研究人员观察到每个个体所携带的变异数量同样随样本大小线性增加,同时罕见变异PRS的性能也随之提升。总之,研究表明可以在不断增加的样本中逐步改善罕见变异PRS的性能。
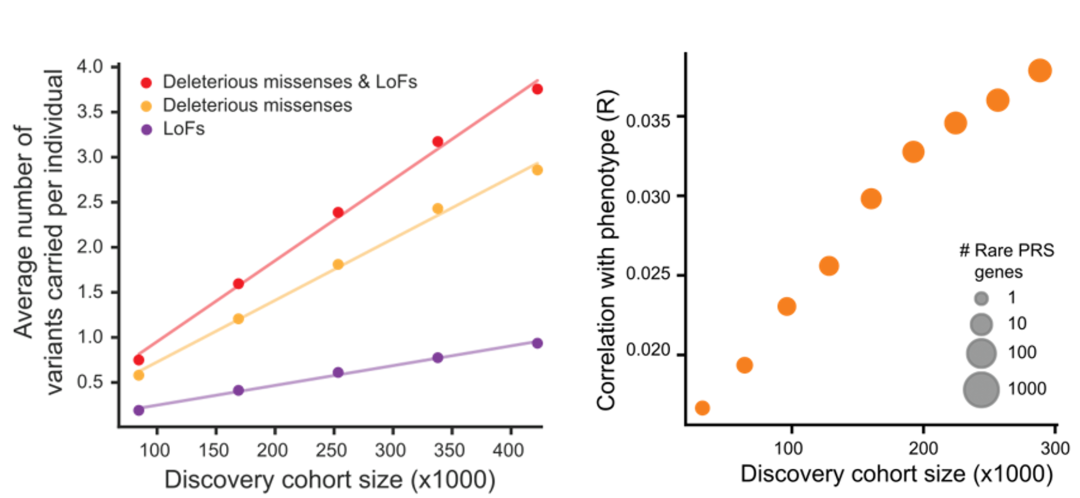
左:每个个体发现的罕见有害变异数量随样本规模线性增加;右:罕见变异PRS的表现随着样本规模的增加而提升
综上所述,深入了解常见疾病中罕见致病性突变的作用,对于精准医疗13-15和靶向药物研发16-18都具有重要意义。在本研究中,利用PrimateAI-3D的最新预测技术,对每种表型相关的罕见突变行建模,揭示了罕见致病性突变在常见人类疾病和复杂性状中的作用。研究证明了常见突变和罕见突变在预测人类疾病风险方面具有互补的效用,常见变异可以解释人群总体变异的较高比例,而罕见变异更容易识别出极高风险的异常个体。最终,罕见变异PRS可以与常见变异PRS相结合,形成一个统一的风险评分模型,从而显著提高常见疾病种高风险个体的识别能力。此外,研究发现该罕见变异PRS模型在不同人群和数据集之间具有更好的迁移性,可以提供比目前以欧洲人为主的遗传研究更普适的结果。
作者信息:illumina人工智能实验室 Kyle Kai-How Farh团队、麻省总医院基因组医学中心Amit V. Khera团队、麻省理工学院和哈佛大学布罗德研究所Anne O'Donnell-Luria团队等为本项目主要研究团队。
参考文献
1. Karczewski K J, Francioli L C, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans[J]. Nature, 2020, 581(7809): 434-443.
2. Gao H, Hamp T, Ede J, et al. The landscape of tolerated genetic variation in humans and primates. Science. In press.
3. Schork N J, Murray S S, Frazer K A, et al. Common vs. rare allele hypotheses for complex diseases[J]. Current opinion in genetics & development, 2009, 19(3): 212-219.
4. Weiner D J, Nadig A, Jagadeesh K A, et al. Polygenic architecture of rare coding variation across 400,000 exomes[J]. medRxiv, 2022: 2022.07. 06.22277335.
5. Marras C, Beck J C, Bower J H, et al. Parkinson’s Foundation P4 Group. Prevalence of Parkinson's disease across North America. NPJ Parkinsons Dis. 2018; 4: 21[J].
6. Wallin M T, Culpepper W J, Campbell J D, et al. The prevalence of MS in the United States: a population-based estimate using health claims data[J]. Neurology, 2019, 92(10): e1029-e1040.
7. Gupta A, Wang Y, Spertus J A, et al. Trends in acute myocardial infarction in young patients and differences by sex and race, 2001 to 2010[J]. Journal of the American College of Cardiology, 2014, 64(4): 337-345.
8. Lawrence J M, Divers J, Isom S, et al. Trends in prevalence of type 1 and type 2 diabetes in children and adolescents in the US, 2001-2017[J]. Jama, 2021, 326(8): 717-727.
9. Khera A V, Chaffin M, Aragam K G, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations[J]. Nature genetics, 2018, 50(9): 1219-1224.
10. Khera A V, Chaffin M, Wade K H, et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood[J]. Cell, 2019, 177(3): 587-596. e9.
11. Nordestgaard B G, Chapman M J, Humphries S E, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society[J]. European heart journal, 2013, 34(45): 3478-3490.
12. Thanabalasingham G, Owen K R. Diagnosis and management of maturity onset diabetes of the young (MODY)[J]. Bmj, 2011, 343.
13. Henderson R, O’Kane M, McGilligan V, et al. The genetics and screening of familial hypercholesterolaemia[J]. Journal of biomedical science, 2016, 23: 1-12.
14. Kuchenbaecker K B, Hopper J L, Barnes D R, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers[J]. Jama, 2017, 317(23): 2402-2416.
15. Cohen S A, Pritchard C C, Jarvik G P. Lynch syndrome: from screening to diagnosis to treatment in the era of modern molecular oncology[J]. Annual review of genomics and human genetics, 2019, 20: 293-307.
16. Sabatine M S. PCSK9 inhibitors: clinical evidence and implementation[J]. Nature reviews cardiology, 2019, 16(3): 155-165.
17. Kaufman B, Shapira-Frommer R, Schmutzler R K, et al. Olaparib monotherapy in patients with advanced cancer and a germ-line BRCA1/2 mutation: An open-label phase II study[J]. 2013.
18. Cannon C P, Blazing M A, Giugliano R P, et al. Ezetimibe added to statin therapy after acute coronary syndromes[J]. New England Journal of Medicine, 2015, 372(25): 2387-2397.








