为什么你构建的进化树会不一致?
1. 进化树的重要性
进化树是我们研究基因、基因组、物种如何演化的基石,也是生物学多个领域研究的重要工具(比如物种分类、物种多样性预测与保护、病原微生物鉴定、生物信息学软件开发)(图1)。
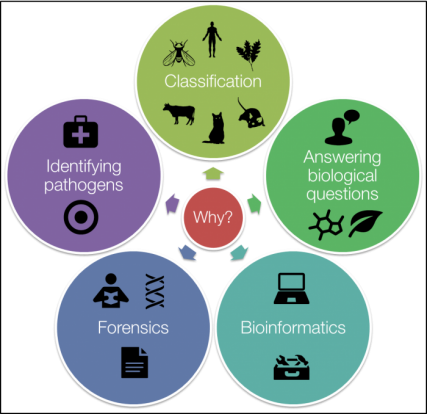
图1 进化树的应用领域
2. 进化树不一致普遍存在
随着基因组测序技术不断进步与成本不断降低,越来越多研究趋向使用大规模组学数据——系统发育基因组学 (Eisen 1998)。这一研究方向极大地帮助我们明确诸多生命之树的重要分支。然而,进化树不一致——相互冲突的进化树——仍然普遍存在 (Rokas and Chatzimanolis 2008),特别是对于生命历史中的关键事件,比如生命之树的根、真核生物的起源、早期动物的起源等(图2)。对于这些冲突的进化树,即便我们用全基因组以及最先进的算法依然不能得到明确的答案。
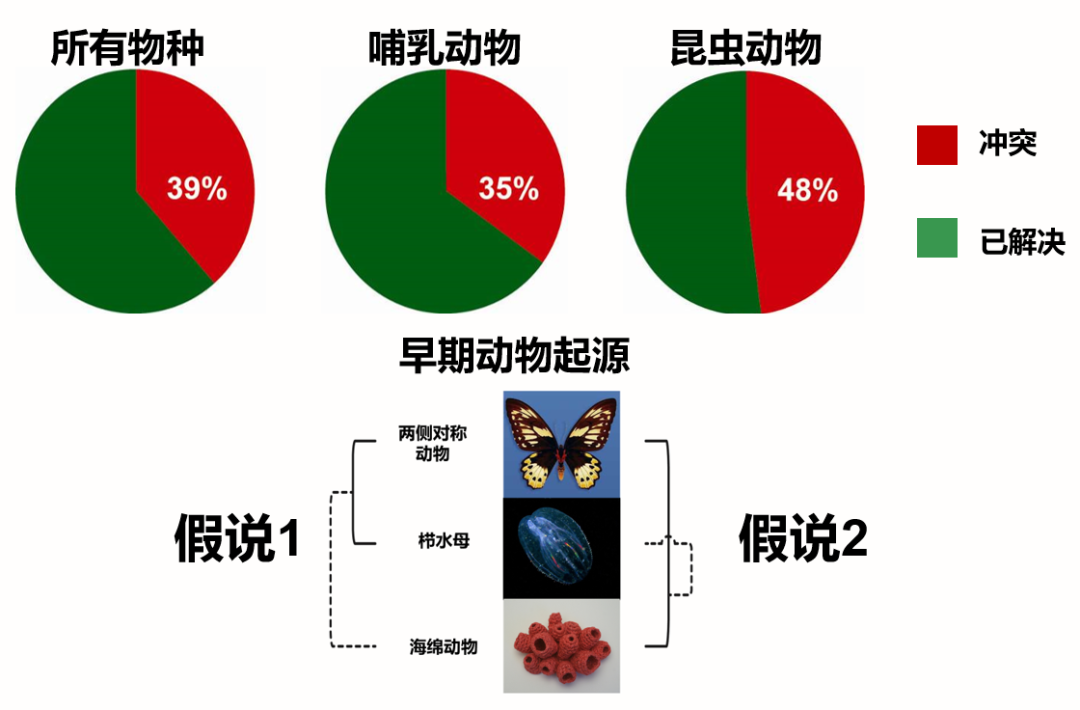
图2 进化树不一致普遍存在
(Rokas and Chatzimanolis 2008)
3. 进化树构建过程
为什么进化树会不一致? 回答这个问题,我们首先需要理清进化树怎么推断出来的。传统进化树的构建是基于形态学数据,目前进化树推断大多数都是基于分子数据(如DNA,蛋白质)。进化树的构建包含多个步骤:物种选择、同源基因鉴定、数据比对、噪音剔除、演化模型选择、进化树构建算法选择等(图3)。从目前大量研究来看,每一步骤均能够导致进化树不一致,只是影响程度存在差异。
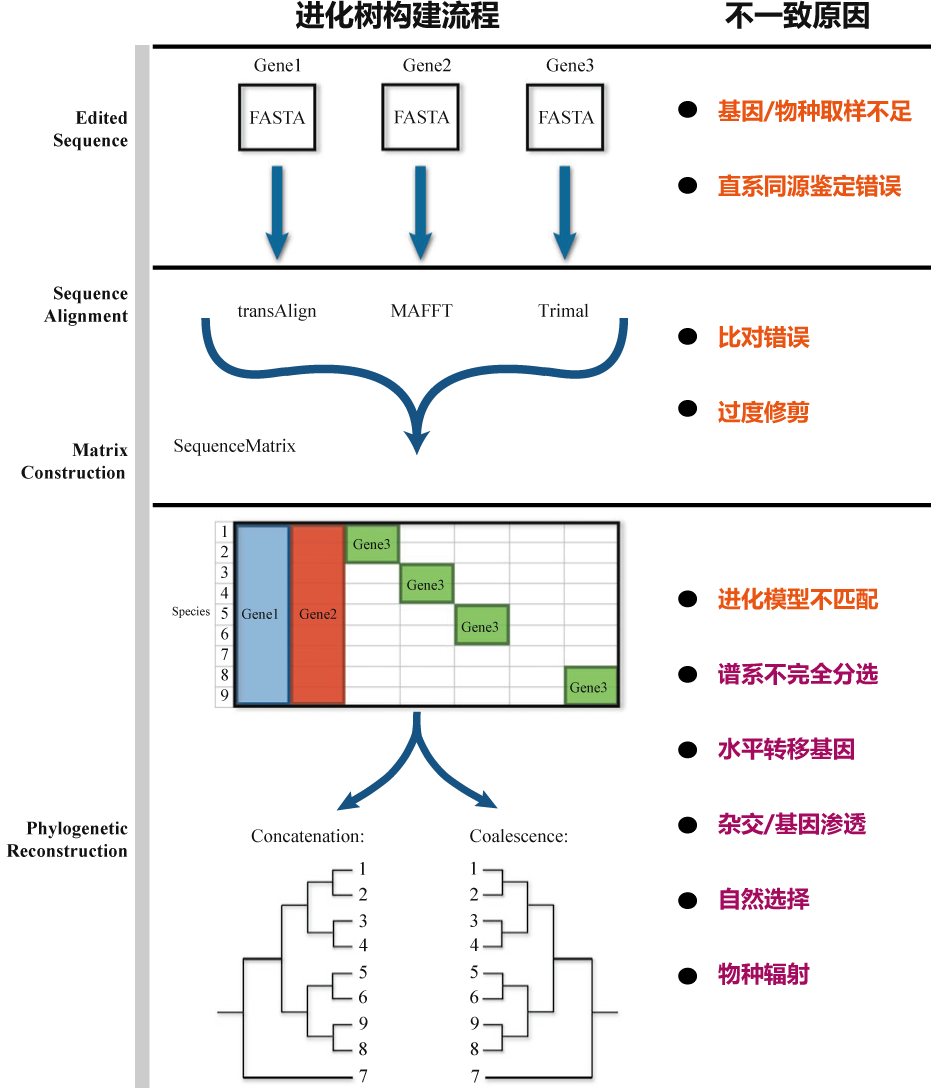
图3 进化树构建过程与不一致原因
4. 常见进化树不一致的原因
常见进化树不一致的原因大致可以分为两大类:分析因素(Analytical factors)和生物因素(Biological factors)(图3和表1)。
分析因素也可以分为随机误差(Stochastic error)和系统误差 (Systematic error)。随机误差主要体现在物种取样量差异、基因或位点数目不足、过度修剪比对矩阵;系统误差主要体现在直系同源基因鉴定的错误、进化模型不匹配。
生物因素主要体现在谱系不完全分选(Incomplete lineage sorting)、水平转移基因(Horizontal gene transfer)、杂交/基因渗透(Hybridization / Introgression)、自然选择(Natural selection)、以及物种辐射(Evolutionary radiation)。

表1 进化树不一致的常见因素总述
5. 其它进化树不一致的原因
除了上述分析因素和生物因素,还有进化树构建软件算法的缺陷。在系统发育基因组学中,基于最大似然法(Maximum Likelihood,简称ML)构建系统发育树已然成为最常用的分析流程之一。最近研究表明最大似然法存在不可重复危机和进化树搜索空间不足现象。
不可重复危机:沈等研究结果表明 (Shen et al. 2020):即使在数据和所有参数完全一致的情况下,IQ-TREE和RAxML-NG软件各自分别存在18.1%和9.3%的树不可重复。比较IQ-TREE和RAxML-NG之间,仅20.3%系统发育树可重复(图4)。
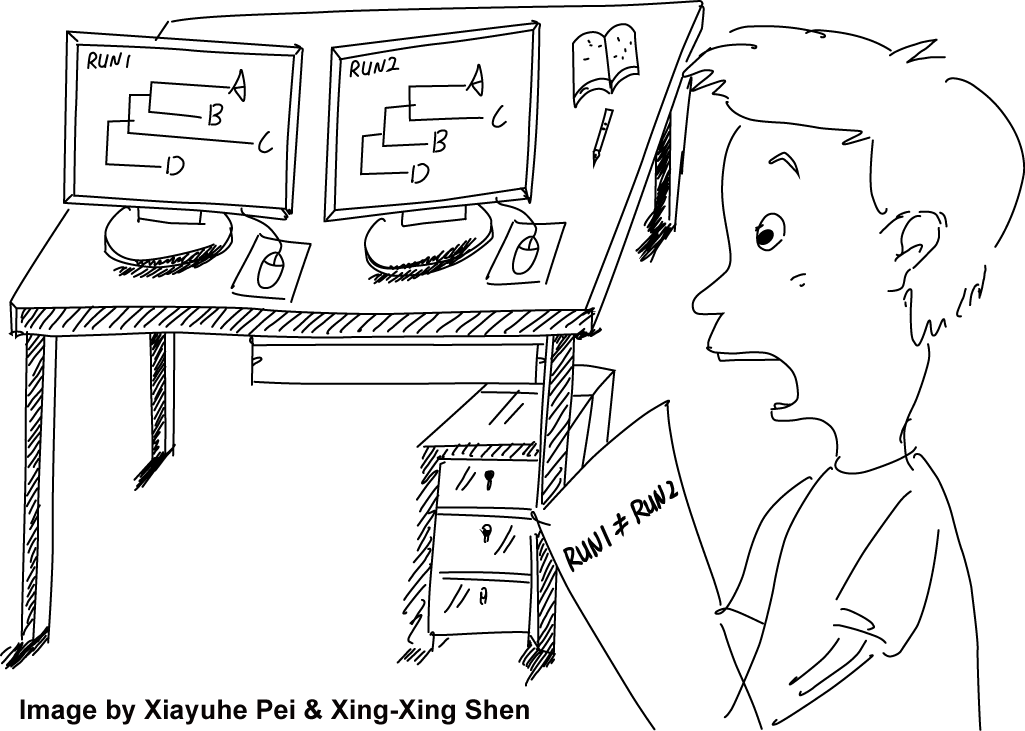
图4 进化树不可重复危机
进化树搜索空间不足:现有ML软件给出的似然树极有可能不是最优的似然树。其主要原因:利用少量搜索次数(常规采用10次以内搜索次数)从海量的树空间中(例如,20个物种就有大约8 x10^21 棵可能有根树)快速地找到得分最高的似然树往往很困难 (Haag et al. 2022)。
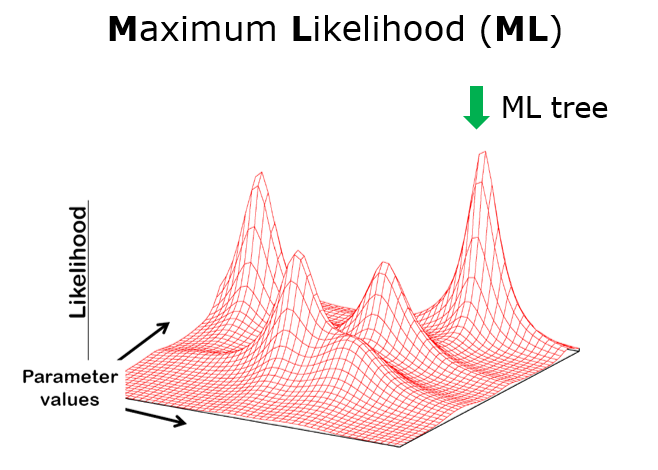
图5 似然树搜素空间庞大
能不能一招制服进化树不一致?
嗯…答案是否。目前没有一个算法或者理论,可以一次性把上述所有不一致原因逐个分析一遍,并给可靠的解决方案。结合近几年系统发育学发展,我们可以尝试如下方案:
1)如果研究是基于少量数据或者小于20个基因,可以尝试解析物种或者基因数目问题(Taxon and gene sampling)。通常从进化枝的支持率(如自展值)可以大致判断出是否由于数据不够导致的。
2)如果研究是基于大规模组学数据,可以尝试解析直系同源基因的鉴定,进化模型不匹配, 谱系不完全分选,以及物种辐射。
除了上述方案,当然还可以尝试估算系统发育信号 (Shen et al. 2017; Zhou et al. 2020)、基因支持频率(Gene support frequencies)(Minh et al. 2020),或者进化树推断异质性估算 (Shen et al. 2021),以便快速找到潜在冲突原因。
如何展示不一致的进化树?
因为进化树不一致是普遍存在的,所以尽可能地包含所有进化树的结果,但可以选择“代表性”进化树作为主要结论进行阐述。
写在最后

图6 选择形态数据还是分子数据来构建进化树?
虽然前面提到进化树可以基于形态数据和分子数据,但是上述讨论进化树不一致的原因都是针对分子数据。因此,我们是不是要换一个角度思考“对于某些进化类群,分子数据(图6里的人)是不是不适合用于进化树的构建?”
参考文献
Eisen J.A. 1998. Phylogenomics: Improving Functional Predictions for Uncharacterized Genes by Evolutionary Analysis. Genome Res. 8:163–167.
Haag J., Höhler D., Bettisworth B., Stamatakis A. 2022. From Easy to Hopeless - Predicting the Difficulty of Phylogenetic Analyses. Mol. Biol. Evol. doi:10.1093/molbev/msac254
Minh B.Q., Hahn M.W., Lanfear R. 2020. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Mol. Biol. Evol. 37:2727–2733.
Rokas A., Chatzimanolis S. 2008. From Gene-Scale to Genome-Scale Phylogenetics: the Data Flood In, but the Challenges Remain. p. 1–12.
Shen X.-X., Hittinger C.T., Rokas A. 2017. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 1:0126.
Shen X.-X., Li Y., Hittinger C.T., Chen X., Rokas A. 2020. An investigation of irreproducibility in maximum likelihood phylogenetic inference. Nat. Commun. 11:6096.
Shen X.-X., Steenwyk J.L., Rokas A. 2021. Dissecting Incongruence between Concatenation- and Quartet-Based Approaches in Phylogenomic Data. Syst. Biol. 70:997–1014.
Zhou X., Lutteropp S., Czech L., Stamatakis A., Looz M. Von, Rokas A. 2020. Quartet-Based Computations of Internode Certainty Provide Robust Measures of Phylogenetic Incongruence. Syst. Biol. 69:308–324.








